探索蛋白自然多样性是理解蛋白功能和蛋白工程改造的关键。环境DNA蕴含海量蛋白序列空间,远超出当前任何数据库容量。利用这些序列需要对专一功能蛋白进行目标性注释而不是广义分类。近日,中国科学院遗传发育所农业资源研究中心李小方研究员团队以“Exploring protein natural diversity in environmental microbiomes with DeepMetagenome”为题,在Cell Reports Methods杂志发了蛋白自然多样性注释方法DeepMetagenome。
DeepMetagenome注释方法主要有三个步骤,包括建立数据库、训练深度学习模型和数据挖掘。所有步骤均有一个Python软件包执行。其深度模型主要包括了Embedding,Conv1D,LSTM和Dense等层。研究建立了一个由1.46亿高质量环境蛋白序列组成的预测数据库,使用DeepMetagenome方法从该数据库中预测到超过500条高置信度金属硫蛋白。研究合成了23条任意挑选的序列,功能实验表明其中的20条展示了预期活性。在对完全不同性质的另外三个蛋白的挖掘中,DeepMetagenome同样展示了高质量预测结果。对所有蛋白的预测对比表明,DeepMetagenome的预测性能远超基于序列的比对方法,也超越了现有的CNN算法模型,在稳定性上甚至超过了强大的Transformer算法。
该方法具有高度的可塑性,广义上可对任意具有特定序列特征的功能蛋白进行预测。这一强大预测能力来源于该方法的几项革新。该方法首先突破了数据库真值的收集问题。常规来讲,基于序列比对和传统深度学习方法的数据库,要求验证功能的真值序列,这大大限制了绝大部分蛋白家族的注释。DeepMetagenome以大部分已注释蛋白序列,尤其是全基因组蛋白序列为真值作为前提,通过特定的生物信息分析步骤剔除错误注释和异常序列,从而建立了具有高置信度的真值序列数据库。方法的另一个创新在于使用了一个具有代表性的阴性序列数据库。传统上,二分类序列注释的阴性序列是手工挑选或随机挑选,这个为阴性序列的覆盖度带来了挑战。DeepMetagenome的阴性序列采用了大肠杆菌泛基因组,以最大程度覆盖非真值序列的其他蛋白家族多样性,这有助于提高二分类注释的准确度。软件的使用非常简单,完成所有步骤仅需四行命令。
本研究得到国家自然科学基金项目的资助(nos. 32250015, U21A2024, and 32101371)。农业资源研究中心李小方研究员为第一作者,郑鑫博士为通讯作者。河北农业大学和中国农业科学院相关研究人员对软件进行了测试和重要补充。
论文链接:https://www.cell.com/cell-reports-methods/fulltext/S2667-2375(24)00286-8
软件包免费下载:https://github.com/uqxli12/DeepMetagenome/blob/main/README.md
交互式代码:https://ars.els-cdn.com/content/image/1-s2.0-S2667237524002868-mmc3.pdf
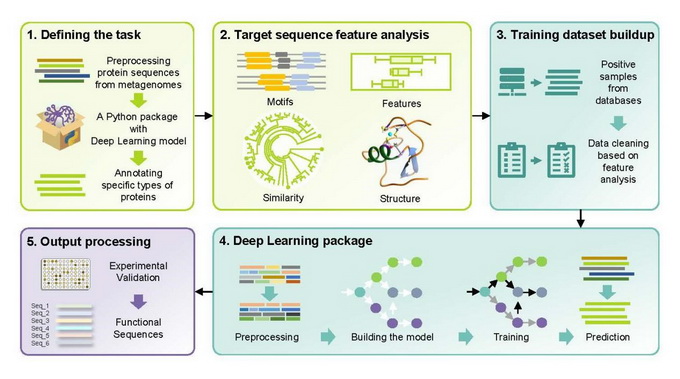
DeepMetagenome方法的基本结构

|
中国科学院遗传与发育生物学研究所农业资源研究中心 京ICP备05002857号-1 地址:河北省石家庄市槐中路286号地理位置与乘车路线 邮编:050022 电话:0311-85814521传真:0311-85815093; Email:zhc@sjziam.ac.cn |

|